
Research Aricle
Austin Chromatogr. 2023; 8(1): 1055.
Simultaneous Determination of Linagliptin and Metformin by Reverse Phase-High Performance Liquid Chromatography Method: An Application in Quantitative Analysis of Pharmaceutical Dosage Forms
Gamze Camlik¹; Fazli Beyazaslan²; Besa Bilakaya¹; Ismail Tuncer Degim¹*
¹Pharmaceutic Technology Department, Faculty of Pharmacy, Biruni University, Istanbul, Turkey
²Nobis Scientific Pharmaceutical Manufacturing, Tuzla, Istanbul
*Corresponding author:Ismail Tuncer Degim Pharmaceutic Technology Department, Faculty of Pharmacy, Biruni University, Istanbul, Turkey. Email: tdegim@biruni.edu.tr
Received: July 31, 2023 Accepted: September 08, 2023 Published: September 15, 2023
Abstract
Linagliptin and Metformin are approved drugs for the treatment of type 2 diabetes mellitus. They are both used in the same dosage forms. We report here that a High-Performance Liquid Chromatography (HPLC) method was developed, optimized, and validated for Linagliptin/Metformin assay. HPLC method is developed for the assay analysis of Linagliptin and Metformin HCl simultaneously; the active substances are 2.5 mg and 1000mg respectively in the film-coated commercial tablet. The concentration range is validated for 0.00025 mg/ml – 0.0025 mg/ml for Linagliptin and 0.1 mg/ml - 1 mg/ml for metformin HCl. Specificity, linearity, precision, intermediate precision, accuracy, and robustness parameters were tested and this proposed method was found to be specific, precise, and robust.
Keywords: Linagliptin; Metformin; HPLC; Validation
Introduction
Linagliptin and metformin are two antidiabetic drugs that are often used in combination for therapy to manage type 2 diabetes [1]. Linagliptin is a Dipeptidyl Peptidase-4 (DPP-4) inhibitor that increases the levels of incretin hormones, stimulating insulin secretion [2]. Metformin is a biguanide that reduces glucose production in the liver and improves insulin sensitivity [3]. Clinical trials have been conducted to evaluate the safety and efficacy of the combination therapy of linagliptin and metformin in patients with type 2 diabetes [4]. In a Phase 2 study, it was found that the combination therapy significantly improved glycemic control compared to either drug alone with no significant adverse events reported [5]. Several phase 3 clinical trials have evaluated the efficacy and safety of linagliptin and metformin combination therapy in patients with type 2 diabetes [6]. In a randomized, double-blind, placebo-controlled trial, the combination of linagliptin and metformin was shown to be superior to either medication alone in reducing HbA1c levels and improving glycemic control over 24 weeks of treatment [7].
However, like any medication, there are potential adverse effects associated with linagliptin and metformin [8]. Some common adverse effects of linagliptin include upper respiratory tract infections, headaches, and hypoglycemia [9]. Common adverse effects of metformin include gastrointestinal symptoms such as nausea, vomiting, and diarrhea [10]. Rare but serious adverse effects of metformin include lactic acidosis, which can be life-threatening [11].
Chromatographic methods have broad applications in both analytical and purification processes [12]. Determining the drug and its byproducts or impurities is crucial, as this information is significant from both the pharmacological and toxicological perspectives, particularly for the pharmaceutical industry and scientists [13]. To ensure safety during pharmaceutical development, it is necessary to establish monitoring techniques for stability tests on raw materials, impurities, and degradation products, due to their potential for toxicity [14]. When the method is straightforward and readily applicable, simultaneous or direct determinations are preferred [14,15]. A chemical component originating from the drug substance or synthesized during the process is considered an impurity in the drug mass [15]. The safety of the drug relies not only on the toxicity of the active substance but also on its pharmaceutical impurities [16]. It is crucial to detect and quantify impurities in raw pharmaceutical materials and degradation products that may form during formulation, manufacturing, or storage. These impurities, also known as "related substances," may have pharmacological or toxicological significance. As such, their presence and levels in products indicate product quality and can pose a risk to patient safety [17-19].
High-Performance Liquid Chromatography (HPLC) is a highly effective analytical method utilized for separating, identifying, and quantifying chemical compounds in diverse sample types [13]. Within pharmaceutical analysis, HPLC is extensively employed for determining drugs and their metabolites in biological fluids, including blood, urine, and plasma, as well as in pharmaceutical formulations [20].
Extensive validation of an HPLC method is essential as it is commonly used to regularly analyze raw materials or drugs during various stages, including material acceptance, preformulation, quality control, and storage [21-23].
We present a rapid and cost-effective HPLC analytical technique to measure the quantities of linagliptin and metformin in pharmaceutical dosage forms or raw materials. The method has been validated by the ICH guidelines on "Validation of Analytical Procedures: Text and Methodology [24].
We evaluated various parameters, including specificity, linearity, recovery, precision (system precision, method precision, and intermediate precision), robustness, solution stability, and system suitability, and found that all the results were within acceptable limits. Therefore, the method can be employed or adopted for other intended purposes.
Materials and Methods
Chemicals
Analytical grade water, Acetonitrile (Isolab, Istanbul, Turkey), Potassium hydroxide (Isolab, Istanbul, Turkey), and Potassium Dihydrogen Phosphate (Merck, Merck KGaA, Darmstadt, Germany) were used.). Other chemicals and ingredients were of analytical grade. Linagliptin/Metformin (2.5/1000) film-coated tablets were obtained from the market.
HPLC
Zorbax SB CN (250mm x 4.6 mm, 5μm) (CA, USA) was used for the separation of compounds in HPLC (Shimadzu, Maryland, USA) analysis with a photodiode array detector. The separation was carried out using an isocratic elution, meaning that the mobile phase composition remained constant throughout the run time. The flow rate was set at 1.0 ml/min, and the column temperature was kept at 25°C. The photodiode array detector was used for the analytical detection of the separated compounds at a wavelength of 265 nm. The run time for the entire separation and analysis process was 8 minutes.
Assay
HPLC method is used for the assay analysis of Linagliptin and Metformin HCl (2.5 mg / 1000mg) in film-coated tablets obtained from the market. The concentration range was 0.00025 mg/ml – 0.0025 mg/ml for Linagliptin and 0.1 mg/ml - 1 mg/ml for metformin HCl. Specificity linearity, precision, intermediate precision, accuracy, and robustness parameters were tested in the report. Phosphate Buffer / Acetonitrile mixture (65:35) was used as a mobile phase. 10μl of solution was injected into the chromatographic system several times (3 times). System compatibility was checked with consecutive runs/readings of the standard solution.
Preparation of Samples from Tablets
10 tablets are weighed. The average weight tablets are crushed and the film tablet powder equivalent to 2.5 mg Linagliptin is weighed into a 200 ml volumetric flask, dissolving by holding it for 10 minutes with the help of an ultrasonic bath and completed with a dilution solution to the volume. 10 ml of this solution is taken to a 100 ml volumetric flask and completed to the volume with dilution solution. It is filtered through a 0.45 μm pore-sized filter and the vial was sealed. (C Linagliptin: 0.00125 mg/mL), (C Metformin HCI: 0.5 mg/mL)
Linagliptin%:
At: Active ingredient peak area obtained from the test solution
As: Active ingredient peak area obtained from the standard solution
Ws: Standard Weighing (mg)
P: as is the potency of standard
Mw: Average weight of tablet (mg)
Wt: Samples weight (mg)
T: Theoretical label content of tablets (2.5 mg)
Metformin HCI%:
At: Active ingredient peak area obtained from the test solution
As: Active ingredient peak area obtained from the standard solution
Ws: Standard Weighing (mg)
P: Potency of standard
Mw: Average weight of tablet (mg)
Wt: Samples weight (mg)
T: Theoretical label content of tablets (1000 mg)
Standard Stock Solution
25 mg of linagliptin working standard is taken into a 200 mL volumetric flask, dissolved by holding it for 10 minutes in the ultrasonic bath, and completed to its volume with dilution solution.
(C Linagliptin : 0,125 mg/mL)
Standard Solution
100 mg of Metformin HCl working standard is taken into a 200 mL volumetric flask, dissolved by holding it for 10 minutes with the help of an ultrasonic bath, and 2 ml of the stock standard solution is added and the volume is completed with the dilution solution. It is filtered through a 0.45 micron RC filter and dialed.
(C Linagliptin : 0.00125 mg/mL), (C Metformin HCI : 0.5 mg/mL)
Placebo Solution:
Approximately 268.5 mg placebo is weighed into a 200 ml volumetric flask, some dilution solution is added, kept in an ultrasonic bath for 30 minutes, and completed with dilution solution to its volume. 10 ml of this solution is taken to a 100 ml volumetric flask and completed to the volume with dilution solution. It is filtered through the 0.45-micron filter and vialled.
Validation Parameters
The Linagliptin/Metformin assay on tablets was performed by ICH guidelines for the validation. The parameters of system suitability, linearity, specificity, recovery, precision, (system, method, and intermediate precision), robustness, and solution stability were tested [16].
Specificity
Specificity is the unequivocal determination of the substance to be analyzed in the product in the presence of matrix effects and additives [13]. The mobile phase, placebo, standard and sample solutions are prepared and analyzed as in the method.
There must be no interference with the active substance from the mobile phase, dilution solution, and placebo. The retention time of Linagliptin is 4.39 minutes and for Metformin HCl is 2.59 minutes.
Linearity
The linearity of the analytical method is that the test results obtained within the specified interval have the correct proportion to the concentration of the relevant substance and/or active substance in the sample. 20%, 50%, 80%, 100%, and 200% standard solutions are prepared for Linagliptin and Metformin HCI, and each concentration is injected and analyzed three times separately. The correlation coefficient was then determined. The correlation coefficient is represented as r2 (it should be >0.995). RSD% value of the peak areas for each concentration should be =1.0. The intercept value should be less than 5% of the average area [13].
Accuracy
The accuracy of the analytical method is expressed as the suitability of the prepared concentration of the substance to be analyzed in the sample and the concentration of the determined result of the assay The accuracy study will be prepared by adding the active substance to the placebo and analyzed as specified in the method, test solutions at 50% 100% and 200% levels and 3 from each level. % Recovery was then calculated. % Recovery should be 98.0% - 102.0%. RSD% of the calculated results from the recovery studies should be =1.0 [13,15].
Precision
The precision of an analytical method is the amount of scattering in the results obtained from multiple analyses of a homogeneous sample [25]. System precisions, repeatability, and intermediate precisions were evaluated.
System Precision
System precision is the measure of system performance independent of errors from the sample preparation phase. 6 consecutive injections are made from the standard solution, and the area values are determined. The mean of the results, Standard Deviation (SD), and percentage Relative Standard Deviation (% RSD) are calculated. The RSD value must not be greater than 2.0%.
Intermediate Precision
To see the effect of random changes during the day in the laboratory on the precision of the analytical method; it is tested by performing the same analysis with different columns and different analysts. Different analysts prepare samples defined with method precision. It is injected into the system using a different HPLC column system. The result is calculated for each sample. The % RSD between the results of 12 samples of two analysts is calculated. The RSD should be =2.0 % (n=12) [13,14].
Repeatability
It is the evaluation of the system performance within the errors that will come from the sample preparation stage [16]. The repeatability of the analytical method for assay analysis is tested using measurement data from 6 original samples prepared separately. Metformin's and Linagliptin's sample concentrations were adjusted to 0.5 mg/ml and 0.001251 mg/ml, respectively. The RSD of the results should be =2.0 %.
Robustness
The robustness of an analytical method is the measure that the Method parameters remain unaffected by known small changes and is an indication of the reliability of the analytical method in its normal use [16]. The following changes will be made in the method parameters and the effects of these changes on the system suitability parameters will be examined.
The Effect of the Temperature
Column temperature will be changed by 5°C (20°C – 25°C – 30 °C). The standard and test solution prepared as described in the sample preparation section is injected into the system and its compliance with the acceptance criteria is checked. The results are calculated and compared with the results obtained under the conditions of the method.
The Effect of Flow Rate
The flow rate will be changed to 0.2 ± ml /min (0.8 ml/min. – 1.0 ml/min. – 1.2 ml/min.). The standard and test solution prepared as described in the sample preparation section is injected into the system and its compliance with the acceptance criteria is checked. The results are calculated and compared with the results obtained under the conditions of the method. The difference between the original method and the modified method results must be =5.0%.
System Suitability
Six consecutive injections were done and the relative standard deviation between consecutive standard injections must be a maximum of 2.0%, the tailing factor should not exceed 1%. The proposed method was also tested for detection and quantification limits. The Limit of Detection (LOD) and Limit of Quantification (LOQ) of the procedure were also calculated according to the 3 s/m and 10 s/m criterion, respectively, where s is the standard deviation of the peak area ratios (n=4) of the sample and m is the slope of the corresponding calibration curve.
Stability of the Working Solutions
The retention period of the substance to be analyzed should be determined stably under the analysis conditions. The holding time to be selected is specific to the analysis. If the active substance degrades within this period and storage conditions, these conditions must be limited and specified in the method. Standard and sample solutions will be prepared according to the method, kept at chromatographic system temperature (5°C) for 24 hours, and analyzed. RSD of the results should be =1.0.
Results and Discussion
The retention time of metformin was 2.608 minutes and it was 4.417 minutes for linagliptin (Figure 1). Different columns with different polarities were tried. The system for standard solutions was accepted as suitable because the relative standard deviation between readings did not exceed 1%, tailing factors were less than 1.8, and the theoretical plate number was higher than 5000.
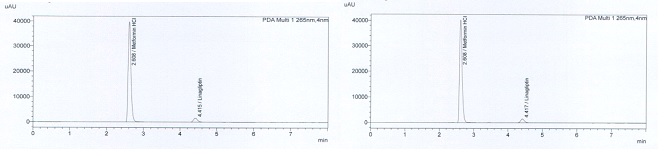
Figure 1: Retention time of analytes (Standards and samples from tablets).
Assay
When commercial 12 tablets were subjected to, the analyses of all tablets from the same producers or from different brand names gave the same result. All tablet contents were found to have an accepted dose (95%-105% of labeled dose) using the proposed method. The method was then subjected to validation analyses.
Specificity
The method was found to be selective for analytes. Mobile phase and diluted solutions were tested and there was no peak around the retention time of the active substances observed on chromatograms. The other peaks at the beginning did not significantly affect the analysis result. Figure 2 shows the chromatograms of diluents and placebo solutions.
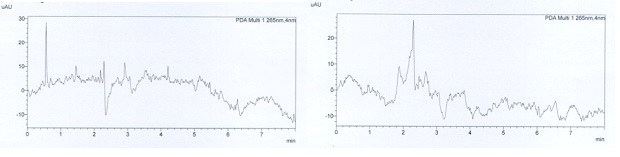
Figure 2: The chromatograms of diluents and placebo solution.
Assay
When commercial 12 tablets were subjected to, the analyses of all tablets from the same producers or from different brand names gave the same result. All tablet contents were found to have an accepted dose (95%-105% of labeled dose) using the proposed method. The method was then subjected to validation analyses.
Specificity
The method was found to be selective for analytes. Mobile phase and diluted solutions were tested and there was no peak around the retention time of the active substances observed on chromatograms. The other peaks at the beginning did not significantly affect the analysis result. Figure 2 shows the chromatograms of diluents and placebo solutions.
Linearity
After 6 consecutive analyses of 5 different concentrations, the correlation coefficient (r2) was higher than 0.995. Linearity study results are summarized in Table 1-2 and Figure 3-4; RSD% values of the areas for all were less than 1.0 and the intercept values were less than 5% of the average area for the 100% linagliptin.
![]()
Theorical Conc. %
Concentration (mg/mL)
Peak Areas
Average Area
SD
RSD%
20,0
0,00025
2338
2321
15,4715
0,67
2306
2308
2309
2340
2324
50,0
0,000625
5646
5656
10,5040
0,19
5656
5667
80,0
0,00100
9164
9152
12,0139
0,13
9153
9140
100,0
0,00125
11617
11611
25,3962
0,22
11617
11633
11584
11577
11639
200,0
0,0025
23266
23284
18,5113
0,08
23309
23276
23305
23270
23276
Table 1: Linearity of Linagliptin.
![]()
Theorical Conc. %
Concentration (mg/mL)
Peak Areas
Average Area
SD
RSD%
20,0
0,10
44988
45013
65,8992
0,15
45009
44926
44983
45119
45050
50,0
0,25
106160
106092
90,1462
0,08
105990
106127
80,0
0,40
168709
168772
58,2838
0,03
168824
168783
100,0
0,50
208396
208516
198,3056
0,10
208467
208916
208411
208467
208436
200,0
1,00
400830
401014
195,7822
0,05
400816
401298
401051
400914
401177
Table 2: Linearity of Metformin.
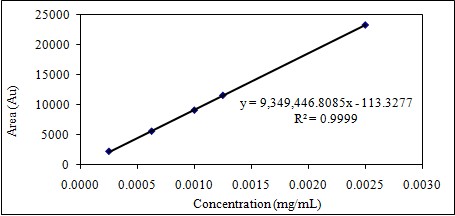
Figure 3: Dose-response graph of Linagliptin.
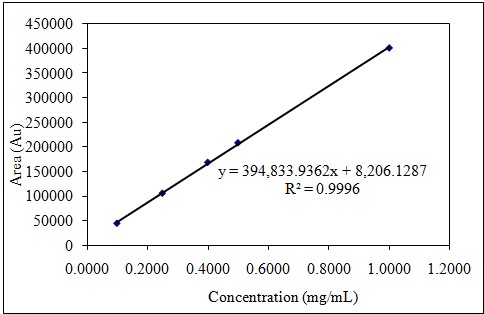
Figure 4: Dose-response graph of Metformin.
Recovery
Recovery studies were carried out in duplicate injections of three different concentrations of standard solutions. Recovery study results are summarized in Tables 3 and 4. Recovery % values were between 95.0% - 105.0% and RSD values were not more than 1.0.
Precision
Intermediate precision, System precision, and repeatability were determined. Mean areas, Standard Deviation (SD), and percentage of the relative standard deviation of 6 consecutive injections of the standard solution (RSD%) were calculated for the system precision (Table 5). Intermediate precision was calculated using the same laboratory, the same column with different serial numbers, and different equipment on different days by different analysts. Six replicates of Linagliptin and Metformin HCL test solutions were prepared and analyzed (Table 6). The repeatability values of six samples from the same batch of Linagliptin and Metformin HCL raw material were analyzed. RSD% for system precision was not more than 1.0 and RSD% for repeatability and intermediate precision was not more than 2.0 (Table 3 & 4).
![]()
Linagliptin 2.5mg Metformin HCI 1000mg Tablets Recovery Study Result
%
Sample
Linagliptin Area
Experimental Conc
Theorical Conc.
% Recovery
Average
RSD%
50
1
5649
0,00062
0,000625
99,88
101,23
0,92
2
5689
0,00063
0,000625
100,58
3
5728
0,00063
0,000625
101,27
4
5775
0,00064
0,000625
102,10
5
5723
0,00063
0,000625
101,18
6
5790
0,00064
0,000625
102,37
100
1
11630
0,00125
0,00125
100,16
99,98
0,19
2
11632
0,00125
0,00125
100,18
3
11577
0,00125
0,00125
99,71
4
11596
0,00125
0,00125
99,87
5
11623
0,00125
0,00125
100,10
6
11595
0,00125
0,00125
99,86
200
1
23329
0,00250
0,0025
100,19
100,50
0,22
2
23346
0,00251
0,0025
100,27
3
23424
0,00252
0,0025
100,60
4
23435
0,00252
0,0025
100,65
5
23458
0,00252
0,0025
100,75
6
23405
0,00251
0,0025
100,52
Table 3: Recovery Results for Linagliptin.
![]()
Linagliptin 2.5mg Metformin HCI 1000mg Tablets Recovery Study Result
%
Sample
Metformin HCI
AreaExperimental Conc.
Theorical Conc.
% Recovery
Average
RSD
50
1
104565
0,24640
0,25
98,56
98,91
0,31
2
104492
0,24623
0,25
98,49
3
105066
0,24758
0,25
99,03
4
105038
0,24752
0,25
99,01
5
105208
0,24792
0,25
99,17
6
105213
0,24793
0,25
99,17
100
1
206287
0,49466
0,50
98,93
99,12
0,18
2
206334
0,49477
0,50
98,95
3
206575
0,49535
0,50
99,07
4
206640
0,49550
0,50
99,10
5
207056
0,49650
0,50
99,30
6
207171
0,49677
0,50
99,35
200
1
398407
0,99350
1,00
99,35
99,54
0,16
2
398254
0,99312
1,00
99,31
3
399430
0,99605
1,00
99,61
4
399567
0,99639
1,00
99,64
5
399529
0,99630
1,00
99,63
6
399788
0,99694
1,00
99,69
Table 4: Recovery Results for Metformin.
![]()
Precision
Sample
Linagliptin
AreaResults %
Metformin HCI
AreaResults %
1
11682
96,02
207871
98,03
2
11507
95,41
204338
97,21
3
11690
96,10
210614
99,35
4
11691
95,57
209039
98,05
5
11809
96,72
212012
99,63
6
11539
95,76
206353
98,26
Average
11653
95,93
208371
98,42
sd
111,63
0,47
2804,34
0,91
rsd
0,96
0,49
1,35
0,92
Table 5: Precision for Linagliptin and Metformin HCL.
![]()
Intermediate Precision
Sample
Linagliptin
AreaResults %
Metformin HCI
AreaResults %
1
11742
96,39
210282
99,05
2
11681
96,12
208516
98,45
3
11771
96,81
210883
99,51
4
11598
96,23
205437
97,80
5
11735
96,17
210668
99,06
6
11819
97,39
210695
99,62
Average
11724
96,52
209414
98,91
SD
76,69
0,49
2132,97
0,69
RSD
0,65
0,51
1,02
0,69
Table 6: Intermediate Precision Results for Linagliptin and Metformin Raw Materials.
Robustness
Column temperature and the flow rate were changed and RSD% of the results was less than 2.0. Table 7 summarizes the results. The difference between the original method and the modified method results must be =5.0%. The change in flow rate and retention times and recovery are reported and the result is evaluated. With the change in the flow rate, changes in Linagliptin and Metformin HCI retention times were observed. The change in recovery values meets the acceptance criteria. The change in flow rate does not adversely affect the results of the analysis. With the change in the column temperature, changes in Linagliptin and Metformin HCI retention times were observed. The change in recovery values meets the acceptance criteria. ± 5 change in column temperature does not adversely affect the results of the analysis.
![]()
0.8 ml/min.
1.0 ml/min
(Validation conditions)1.2 ml/min
Analyte
Retention Time (min)
%Recovery
Retention Time (min)
%Recovery
Retention Time (min)
%Recovery
Linagliptin
5,53
96,84 %
4,44
96,90 %
3,71
96,71 %
Metformin HCI
3,25
98,61 %
2,61
98,91 %
2,18
98,60 %
Table 7: The effect of flow rate on the analyses.
The Limit of Detection (LOD) and Limit of Quantification (LOQ) of the procedure were also calculated as 3.56 ng and 35.6 ng for Linagliptin where these were 0.466 μg and 4.66 μg for metformin.
![]()
20C
25C
(Validation conditions)30C
Analyte
Retention Time (min)
%Recovery
Retention Time (min)
%Recovery
Retention Time (min)
%Recovery
Linagliptin
4,39
96,30 %
4,44
96,90 %
4,49
96,82%
Metformin HCI
2,59
98,32 %
2,61
98,91 %
2,63
98,35%
Table 8: The effect of temperature on the analyses.
Stability of the Working Solution
Standard and test solutions were prepared and tested at room temperature for at least 24 hours and analyzed. RSD% of the results was not more than 1.0. Solution stability test results are summarized in Table 9. The Standard and sample solution was found to be stable at 5°C after 24 hours.
![]()
Solution
Initial Area
(T0)Area
(T24)Correlation %
Linagliptin Standard
12113
12133
100,17%
Linagliptin Sample
11761
11795
100,29%
Metformin HCI Standard
209036
209021
99,99%
Metformin HCI Sample
211283
210547
99,65%
Table 9: Stability of standard solutions at room temperature.
Conclusion
The developed method was found to be specific to Linagliptin and Metformin HCL. The mobile phase and diluted solutions were not representing any significant interfering peak. The correlation coefficient was r2=0.9998, and recovery was 100.03%. RSD% was 0.23, and 0.05 for system precision, repeatability, and reproducibility respectively. A little change in parameters such as oven temperature and flow rate did not affect the results. The standard solution was found to be stable at 25°C for 24 hours. RSD% was 0.01. RSD% for system suitability was 0.23. Finally, the developed method is fully validated and can be used as a cheap, easy, and time-saving method for the analysis of Linagliptin and Metformin HCL raw material. This proposed method can be easily adapted for Linagliptin and Metformin HCL analyses in pharmaceutical products or solutions or biological samples.
Author Statements
Conflict of Interest Statement
The authors declare that there is no conflict of interest.
References
- Lajara R. Use of the dipeptidyl peptidase-4 inhibitor linagliptin in combination therapy for type 2 diabetes. Expert Opin Pharmacother. 2012; 13: 2663-71.
- Barnett AH. Linagliptin: a novel dipeptidyl peptidase 4 inhibitor with a unique place in therapy. Adv Ther. 2011; 28: 447-59.
- Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003; 29: 6S28–35.
- Haak T. Combination of linagliptin and metformin for the treatment of patients with Type 2 diabetes. Clin Med Insights Endocrinol Diabetes. 2015; 8: 1-6.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE, et al. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with Type 2 diabetes. Diabetes Care. 2006; 29: 2632-7.
- Lv Q, Shen J, Miao L, Ye B, Schepers C, Plat A, et al. Early combination therapy with linagliptin and metformin in people with Type 2 diabetes improves glycemic control to HbA1c = 6.5% without increasing hypoglycemia: pooled analysis of two randomized clinical trials. Diabetes Ther. 2020; 11: 1317-30.
- Gallwitz B, Rosenstock J, Rauch T, Bhattacharya S, Patel S, von Eynatten M, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomized, double-blind, non-inferiority trial. Lancet. 2012; 380: 475-83.
- Scheen AJ. Efficacy and safety of Jentadueto® (linagliptin plus metformin). Expert Opin Drug Saf. 2013; 12: 275-89.
- Sortino MA, Sinagra T, Canonico PL. Linagliptin: A thorough Characterization beyond Its Clinical Efficacy. Front Endocrinol (Lausanne). 2013; 4: 16.
- Siavash M, Tabbakhian M, Sabzghabaee AM, Razavi N. Severity of gastrointestinal Side Effects of metformin Tablet Compared to metformin Capsule in type 2 diabetes mellitus Patients. J Res Pharm Pract. 2017; 6: 73-6.
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycemia in Type 2 diabetes: A patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012; 35: 1364-79.
- Christian Ebere E, Obinna Isiuku B, Andrew Wirnkor V. Applications of column [paper]. Thin layer and ion exchange chromatography in purifying samples: mini review. SF J. Pharm Anal Chem. 2019; 2: 1-6.
- Camlik G, Beyazaslan F, Kara E, Ulker D, Albayrak I, Degim IT. A validated high-pressure liquid chromatography (HPLC) method for linagliptin and metformin HCl. Med Res Arch. 2022; 10: 2022.
- Akay C, Degim IT, Sayal A, Aydin A, Özkan Y, Gül H. Rapid and simultaneous determination of acetylsalicylic acid, paracetamol, and their degradation and toxic impurity products by HPLC in pharmaceutical dosage forms. Turk J Med Sci. 2008; 38: 167-73.
- Degim T, Akay C, Büyükafsar K, Cevheroglu S. Simultaneous determination of codeine and ethyl morphine HCL in tablet formulations using LC. J Pharm Biomed Anal. 2001; 26: 15-21.
- Kowalska M, Wozniak M, Kijek M, Mitrosz P, Szakiel J, Turek P. Management of validation of HPLC method for determination of acetylsalicylic acid impurities in a new pharmaceutical product. Sci Rep. 2022; 12: 1.
- Guideline IHT. Impurities in new drug substances. Q3A, In an International conference of harmonization 2008.
- Guideline IHT. Impurities in new drug products. Q3B (R2), current step. 2006; 4: 1-5.
- Liu DQ, Wu L, Sun M, MacGregor PA. On-line H/D exchange LC–MS strategy for structural elucidation of pharmaceutical impurities. J Pharm Biomed Anal 2007; 44: 320–9.
- Uslu B, Özkan SA. Determination of Lamivudine and Zidovudine in binary mixtures using first derivative spectrophotometric, first derivative of the ratio-spectra and high-performance liquid chromatography–UV methods. Anal Chim Acta. 2002; 466: 175-85.
- Ozkan SA, Uslu B. Rapid HPLC assay for lamivudine in pharmaceuticals and human serum. J Liq Chromatogr Relat Technol. 2002; 25: 1447-56.
- Pham-Huy C, Stathoulopoulou F, Sandouk P, Scherrmann JM, Palombo S, Girre C. Rapid determination of valaciclovir and acyclovir in human biological fluids by high-performance liquid chromatography using isocratic elution. J Chromatogr B Biomed Sci Appl. 1999; 732: 47-53.
- Guideline IHT. Validation of analytical procedures Q2. In: International conference of harmonization. Geneva, Switzerland. 2022; R1.
- AylinHacioglu SevgiKarakus A. Development and validation of HPLC method for determination of nateglinide in drug substances. 2015; 19: 103-8.