
Research Article
Austin J Biotechnol Bioeng. 2024; 11(1): 1128.
PBPK Modeling Characterizes the Impact of Administration Form and Drug-Drug Interaction on Aconitine
Yue Gao1,2*; Yun-xuan Ge 1,2#; Zhuo Zhang2#; Xiao-Mei Zhuang3; Jia-Yi Yan2; Jing Gao2; Zeng-chun Ma2; Yu-guang Wang2
¹Faculty of Environment and Life, Beijing University of Technology, China
²Department of Pharmacology and Toxicology, Beijing Institute of Radiation Medicine, China
³State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, China
*Corresponding author: Yue Gao Faculty of Environment and Life, Beijing University of Technology, No. 100 Pingleyuan, Beijing 100124, China. Tel: +86 010 66931312 Email: gaoyue@nic.bmi.ac.cn
#These authors equally contributed this article
Received: January 11, 2024 Accepted: February 13, 2024 Published: February 20, 2024
Abstract
Aconitine (AC), the main efficient and toxic components of FUZI, has been studied extensively, but the investigation of appropriate in vivo pharmacokinetic model are limited. The aim of this study was to develop Physiologically Based Pharmacokinetic models (PBPK) to predict organ exposure in different doses and methods of administration. Moreover, Paeoniflorin (PAE) and Glycyrrhetinic Acid (GA) were selected to develop a Drug-Drug Interaction (DDI) model with AC. The developed AC-PBPK and relative DDI model of multiple perpetrator drugs was able to adequately describe plasma concentrations and tissue distribution in rats. Furthermore, drug exposure by different administration forms were compared. The effects of drug compatibility on the pharmacokinetics of AC in rats were revealed. This study constructed accurate AC-PBPK and its relative DDI model for the first time, which is suitable for rats with multi-doses and routes of administration. It can be extrapolated to predict the in vivo exposure of AC, solve the problem that the drug concentration in tissue is difficult to obtain, which is applied to clarify the ADME process and toxicity mechanism of AC.
Keywords: Aconitine; Physiologically based pharmacokinetic model; Drug-drug Interaction; Tissue distribution.
Introduction
Aconiti Lateralis Radix Praeparata (FUZI), is one of the famous traditional Chinese medicine, which known as "the primary medicine to restore yang and save reverse". The clinical application of FUZI in traditional Chinese medicine has a history of thousands of years and was mainly used for the deficiency of vital energy and the exhaustion of pulse. Aconitine (AC), is mainly pharmacologically active compounds and toxic ingredients of Aconiti Lateralis Radix Praeparata (FUZI). As the quality control components of FUZI, AC is closely related to the toxicity and efficacy of FUZI related preparations. For instance, according to the announcement on the catalogue of several drug standards issued by the China Food and drug administration (CFDA, Ws3-b - 3427-98-2013 Shenfu injection), the content of AC in Shenfu injection should be regulated at 0.1mg/ml to maintain the balance between efficacy and toxicity. However, the strong cardiotoxicity and neurotoxicity of AC make it severely resistant in clinical therapy, meanwhile, hepatotoxicity has been reported frequently. Scholars all over the world have studied the toxicity mechanism of AC. Wang et al. found aconitine induce apoptosis through mitochondrial-mediated and death receptor signaling pathways in HT22 cells [1]. Qing Xia discussed the developmental toxicity in zebrafish embryo/larvae induced by AC [2]. AC can prolong cardiac QT interval and induce liver cell apoptosis [3], resulting in cardiac and liver toxicity. With a narrow therapeutic index, serious toxic effect may occur even if the oral dose of AC is a little bit higher than the therapeutic dose. Existing research focused on the toxicological mechanisms of AC, but limited data are available on its Absorption, Distribution, Metabolism and Excretion (ADME) processes. Besides, the relationship between the change of in vivo concentration and compatibility detoxification was rarely reported. AC are mainly transported by P-glycoprotein (P-gp) [4]. P-gp is expressed in small intestinal epithelium, heart, liver and brain, which hinders in vivo absorption of AC. Cytochrome P450 enzyme (CYP450) is involved in the metabolism of diester Aconitum alkaloids in human liver [5]. As the metabolic substrate of CYP3A, AC is mainly metabolized by CYP3A1/2 in rats, which could decrease the toxicity after metabolism [6,7]. Licorice and Chinese herbaceous peony impact on the expression of metabolic enzymes, mainly CYP3A1/2 in rat liver [8,9], and accelerate the metabolism of AC in rat liver microsomes, which reduce the toxicity of AC [10,11]. Glycyrrhetinic Acid (GA) and Paeoniflorin (PAE), the main component of licorice and Chinese herbaceous peony, can also affect the expression of CYP450 enzyme and P-gp [9,12]. Above all, we hypothesis the reduction exposure of AC may be induced by GA and PAE through CYP3A and P-gp.
Lack of effective methods to predict toxicity limited the application of AC. In recent years, Physiologically Based Pharmacokinetic (PBPK) model has been widely concerned for a powerful tool to quantitatively delineate how certain extrinsic and intrinsic factors might influence the nonproportional systemic exposures [13,14]. Because it can accurately predict the ADME process and organ exposure based on drug-dependent physicochemical and PK parameters as well as drug-independent physiologic systemic parameters, PBPK model is widely used in the prediction of new Drug-Drug Interaction (DDI) [15,16]. However, there are few reports on the prediction of target organ exposure of toxic components of traditional Chinese medicine by PBPK model. This study intends to establish a PBPK model approach to predict AC exposures in rats after various routes of administration and the effect of GA and PAE on the in vivo concentration of AC. To achieve this, key in vitro and in vivo paraments for AC were collected. Then a bottom-up combined with top-down PBPK model was developed and applied in the assessment of linear PK in rats, at least from the range of 0.5mg/ kg to 1.5mg/ kg. Finally, a DDI-PBPK model was built to predict the effect of DDI drug on the in vivo process of AC.
Materials and Methods
Simulation of PBPK Models
The physicochemical and biopharmaceutical parameters required for drug modeling were obtained by consulting the literature. The missing parameters are predicted by ADMET predictor (Table 1). Import the modeled compound structure into gastro plus software (simulations plus, Inc., California, United States), and input the parameters obtained from literature, predicted parameters and relevant pharmacokinetic parameters obtained from experiments (including metabolism, transport related parameters and measured tissue/plasma partition coefficient) into the software. Advanced Atrioventricular Absorption and Transport (ACAT) model was used to simulate the concentration time curve, and the individual PBPK models of each compound were established respectively. The established rat PBPK model was further optimized by the observed values and work flow was listed as (Figure 1).
![]()
Parameter
Value
Source (PMID)
Molecular weight (g/mol)
645.75
Measured in this study
Log P
0.3
Pubchem
Solubility at pH 9.32(mg/ml)
0.76
Gastro Plus 9.8
Papp(cm/s*10^5)
0.076
23200901
Fu,p
0.72
EPA DataBase
Rbp
0.72
Gastro Plus 9.8
pKa
5.88
CAS DataBase
Table 1.1: Physicochemical and PK parameters of AC used for PBPK models.
![]()
Parameter
Value
Source (PMID)
Molecular weight (g/mol)
480.47
Measured in this study
Log P
-0.19
Pubchem
Solubility at pH 7(mg/ml)
50.57
Gastro Plus 9.8
Papp(cm/s*10^5)
0.07
Drug bank
Fu,p
0.55
EPA DataBase
Rbp
0.74
Gastro Plus 9.8
pKa
5.88
Gastro Plus 9.8
Table 1.2: Physicochemical and PK parameters of PAE used for PBPK models.
![]()
Parameter
Value
Source (PMID)
Molecular weight (g/mol)
470.7
Measured in this study
Log P
6.4
Pubchem
Solubility at pH 9.32(mg/ml)
0.00632
Gastro Plus 9.8
Papp(cm/s*10^5)
0.2
Drug bank
Fu,p
1*10-8
EPA DataBase
Rbp
0.74
Gastro Plus 9.8
pKa
5.1
CAS DataBase
Table 1.3: Physicochemical and PK parameters of GA used for PBPK models.
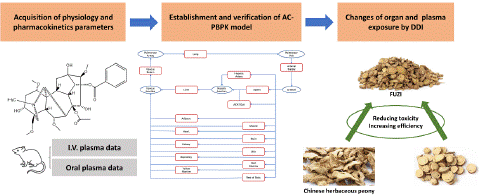
Figure 1: Schematic of permeability-limited tissue model.
Volume of Distribution and Tissue to Plasma Partition Coefficient
The permeability-limited tissue model (Figure 2) was selected for the model optimization by Analysis of AC properties. Steady state volume of distribution (Vss) is estimated according to Poulin & Theil-Extracellular, as described by Eq.1. Initial Vss and Kp value (Table 2) were further optimized by the observed PK data.
![]()
Value
Unit
Source
CLint
0.18
L/h
Calculated
Kplung
4.52
-
Optimized
Kpadipose
4.23
-
Optimized
Kpmuscle
4.29
-
Optimized
Kpliver
4.29
-
Optimized
Kpspleen
4.29
-
Optimized
Kpheart
4.41
-
Optimized
Kpbrain
4.23
-
Optimized
Kpkidney
4.35
-
Optimized
Kpskin
4.64
-
Optimized
KpreproOrg
4.41
-
Optimized
Kpred marrow
4.41
-
Optimized
Kpyellow marrow
4.23
-
Optimized
Kprest of body
4.35
-
Optimized
Table 2: Optimized Kp value of AC in rats.
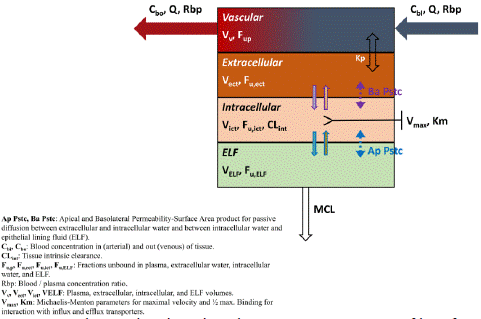
Figure 2: Observed and predicted concentration–time profiles of Aconitine in rats after intravenous and oral administration. Red points represent the concentrations obtained in rats; Black curves describe the simulated concentration–time profiles with AC-PBPK model.
Where Vp is volume of plasma, Ve represents erythrocyte volume, E: P is erythrocyte to plasma concentration ratio (calculated from blood/plasma concentration ratio and hematocrit), Vt represents tissue volume and ERt is the extraction ratio for a given tissue.
Simulation of DDI Models
DDI modules were added to the established AC-PBPK models. After import the established PBPK models of PAE and GA, respectively, select AC, as victim (substrate), PAE and GA as perpetrator in Dynamic Simulation, input the relative CYP3A1/2 and P-gp parameters to simulate the established DDI.
Statistical analysis and model evaluation
For accuracy assessment of a model simulation, acceptance is set to be within 2-fold. For comparison between groups, the statistically significant difference is set for a P value of less than 0.05 using analysis of variance, with the student’s t-test.
Results
Oral and Intravenous Administration of AC-PBPK Model
Based on the physicochemical properties and pharmacokinetic parameters (intravenous administration: 0.04mg/ kg; oral administration: 0.504mg/ kg) [17] of AC, Intravenous PBPK model was established firstly (Figure 3A). Under the same related parameters, oral PBPK model was further constructed and optimized (Figure 3B). The observed value of 1.5mg/kg oral administration to rats was used for accuracy verification of oral PBPK model. In comparation with the simulated value, the ratios of main PK parameters (Cmax, Tmax and AUC) were less than 2 times (Table 3), indicating that the established model was accurate and reliable, and could be used for the further prediction of AC pharmacokinetic behavior in rats.
![]()
Label
Simulated
Observed
Fold error (S/ O)
Units
Cmax
31.47
26.2±5.19
1.20
ng/ml
Tmax
1.04
1.33±0.26
0.78
h
AUC(0-t)
127.86
161±37.8
0.79
ng-h/ml
AUC(0-8)
128.15
167±38.4
0.77
ng-h/ml
Table 3: Predicted and observed PK parameters in rat treated by oral administration of AC (1.5mg/ kg).
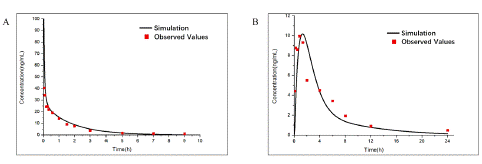
Figure 3: Observed and predicted concentration–time profiles of Aconitine in rats after different multiple administration intervals. Red points represent the concentrations obtained in rats and black curve describes the simulated concentration–time profiles with AC-PBPK model in Figure. 3A. Red curve represents the simulated concentration–time profiles after postprandial administration; Black and blue curve represents the simulated concentration–time profiles after every 24 hours 0.504mg/ kg and 1.51 mg/ kg oral administration in Figure. 3B, respectively.
Comparison of Plasma Concentration of AC by Different Multiple Administration Intervals
After the accuracy verification of multiple oral administration (oral administration: 0.504mg/ kg) [17] AC-model was completed (Figure 4A), the difference of AC plasma concentration between postprandial administration (oral administration: 0.504mg/ kg; The administration intervals were 4, 6 and 14 hours per day) and 24-hour interval administration (oral administration: 0.504mg/ kg and 1.51mg/ kg), which the most commonly administration forms, were compared (Figure 4B). Due to the rapid in vivo clearance, AC concentration of 24-hour interval administration (oral administration: 0.504mg/ kg) fell back to a lower level (0.02ng/ ml). By contrast, postprandial administration can maintain a higher AC minimum concentration (0.4ng/ ml) and the narrower range of plasma concentration compared with the same total dosage of 24-hour interval administration (oral administration: 1.51mg/ kg), which indicated a more effective and safe administration regimen.
Figure 4: Observed and predicted concentration–time profiles of Paeoniflorin and glycyrrhetinic acid in rats after oral administration. Red points represent the concentrations obtained in rats; Black curves describe the simulated concentration–time profiles with PAE-PBPK model and GA-PBPK model, respectively.
Oral Administration of DDI Drug-PBPK Model
PAE and GA were used to investigate the effect of DDI for AC. Based on the physicochemical properties and pharmacokinetic parameters of PAE (oral administration: 50mg/ kg, published in Acta Universitatis Medicinalis Anhui, 2009,44(06) China) and GA (oral administration: 15mg/ kg) [18] related oral PBPK models was established (Figure 5), which precise simulated pharmacokinetic in rats.
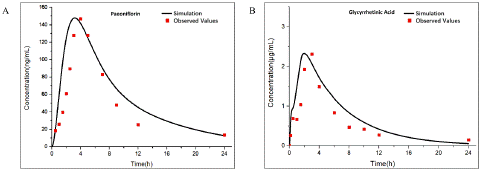
Figure 5: Observed and predicted concentration–time profiles of Aconitine in rats after Paeoniflorin combined administration. Blue and red points represent the concentrations obtained in rats before and after combined administration respectively; Blue and red curves describe the simulated concentration–time profiles before and after combined administration, respectively.
DDI-PBPK Prediction of AC
The established PAE (Figure 6) and GA (Figure 7) PBPK models were added in AC-PBPK model to detect the effect of DDI on the pharmacokinetics of AC (Table 4.). oral administration regimens with 0.2mg/ kg of AC and 20mg/ kg of PAE were used for AC-PAE DDI model [19]. Besides, AC-GA DDI model was established based on oral administration regimens with 1mg/ kg of AC and 10mg/ kg of GA [20] Less 2-fold errors compared with observed value of plasma concentration, the accuracy of DDI model was verified. After that, the established D DI-model was used to simulate the changes of PK in heart, liver and kidney under same oral administration conditions, The simulated results exhibited that PAE and GA have different drug interactions with AC, which reduce or increase the exposure of AC to plasma and organs in in vivo.
![]()
Parameters
AC
AC+PAE
Units
Cmax
6.40
3.10
ng/ml
AUC(0-8)
26.54
12.69
ng-h/ml
Table 4.1: Predicted AC-PK parameters in rats combined administration of PAE.
![]()
Parameters
AC
AC+GA
Units
Cmax
4.70
5.63
ng/ml
AUC(0-8)
23.75
24.29
ng-h/ml
Table 4.2: Predicted AC-PK parameters in rats combined administration of GA.
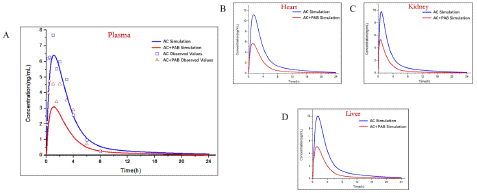
Figure 6: Observed and predicted concentration–time profiles of Aconitine in rats after glycyrrhetinic acid combined administration. Blue and red points represent the concentrations obtained in rats before and after combined administration respectively; Blue and red curves describe the simulated concentration–time profiles before and after combined administration, respectively.and after combined administration, respectively.
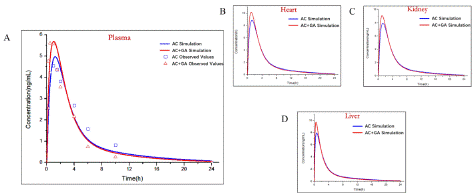
Figure 7: Observed and predicted concentration–time profiles of Aconitine in rats by perfusion-limited and permeability-limited tissue model. Red points represent the concentrations obtained in rats; Green and blue curves describe the simulated concentration–time profiles by perfusion-limited and permeability-limited tissue model, respectively.
Discussion
FUZI is a famous traditional Chinese medicine which is characterized by strong anti-inflammatory, analgesic and other pharmacological effects, commonly used in the clinical treatment of rheumatic and cardiovascular diseases. However, several adverse and poisoning reactions which is related to the strong toxicity of diester-diterpene type Aconitum alkaloids limit its clinical application. As the most significant type of Aconitum alkaloids, AC has been widely studied. But the investigation of appropriate in vivo PK model is limited. The combined therapy of FUZI and licorice or Chinese herbaceous peony is used for reducing toxicity and increasing efficiency, yet the rationality of the compatibility of the two drugs has not been clarified. Although glycyrrhizic acid is the most active components of licorice, it would be hydrolyzed by intestine and transformed into GA after oral administration. Therefore, the PBPK model of AC and the DDI model with GA and PAE were established to dynamically predict the plasma and organs exposure of AC by multiple administration forms and the effect of DDI drug.
In the process of model establishment, PB and PK parameters collection is worth prioritizing and accomplishing for the determination of the general framework of AC-model. Small molecule and its specific solubility determined the feasibility of AC-model development. The distribution volume and clearance rate which related to the variation characteristics of drug plasma concentration were calculated from the I.V. and oral PK data. Besides, the low bioavailability of AC reported from literature was only 1.3-8.2% [17,21], so the first pass effect was adjusted to match the actual curve. The right determination of perfusion-limited or permeability-limited tissue model is another key to successful model establishment. If the permeability of a drug is high, the amount of drug that partitions into the tissue will be limited by the blood flow rate (perfusion rate) through the tissue. If the drug permeability is low, the amount of drug that partitions from plasma into the tissue is limited by the permeability and the surface area available for permeation and/or is governed by saturable transport mechanisms. Moreover, permeability-limited model drug is characterized by carrier-mediated drug transport via influx and efflux transporters. The logP of AC is 0.3 (Computed by XLogP3 3.0) with a low permeability [22]. The existing data showed that the active efflux of AC across Caco-2 cells was mediated mainly by P-gp [4,23], AC increased P-gp expression and transport activity significantly [24]. Thus, permeability-limited tissue model was chosen for the further simulation. In the modeling process, we also developed the AC-PBPK model with perfusion-limited tissue model (Figure 8). The results indicated that permeability-limited tissue model is suitable, while the wrong distribution volume was calculated by perfusion-limited tissue model. Furthermore, the suitable limited tissue mode was selected in development process of GA and PAE PBPK models, ensure that application models can accurately simulate the in vivo exposure of GA and PAE.
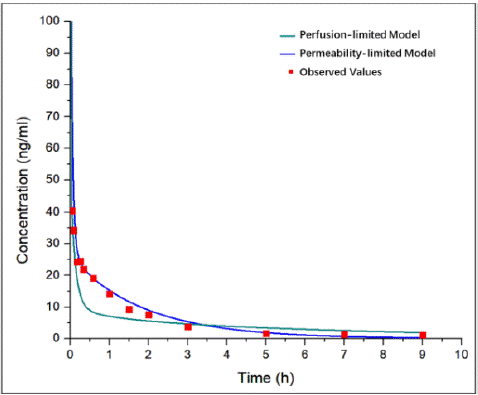
Figure 8:
Licorice and Chinese herbaceous peony were frequently used in traditional Chinese medicine clinical therapy with FUZI for toxicity reduction. For licorice, Glycyrrhizic acid and GA are both main components and Glycyrrhizic acid is hydrolyzed into GA in in vivo process. The similar chemical structure with steroids of GA leads to a corticosteroid like effect [25]. For Chinese herbaceous peony, PAE is the major active component and exhibited extensive anti-inflammatory and immune regulatory effects [26]. Therefore, GA and PAE are added to the model of victim drugs, the model of perpetrator drugs is also introduced. The selected dosing regimens were estimated from the ratio of recommended clinical dosage and the stipulated levels of respective herbs according to the Chinese Pharmacopoeia (Edition 2020). The existing data suggested that AC is the metabolic substrate of CYP3A [5]. The efflux rate of AC is between 23.87 and 55.34(27), which means AC is potential substrate of P-gp [20]. The in vitro parameters data of P-gp and CYP3A1/2 were introduced to predict DDI. After the predicted plasma PK curve which obtained from optimized DDI model and the measured value accord, the AC concentration in liver, kidney and heart, the major metabolic and targeted organs, were simulated. By the comparation of the pharmacokinetic date after single administration and combined administration, it was indicated that GA and PAE affect the in vivo plasma and tissue exposure of AC by different effects on CYP3A1/2 and P-gp activities. PAE decrease the systemic exposure of AC through upregulated the activity of CYP3A and P-gp, so as to reduce the toxicity of AC, which revealed one possible way of compatibility detoxification. Different with PAE, GA inhibited CYP3A and upregulated P-gp expression, inducing a slightly rising Cmax and AUC after combined administration, which indicated other possible toxicity reduction effects existed of GA.
With the optimized DDI models, the in vivo exposure of AC with multiple doses or combined with other drugs can be accurate predicted. After selected suitable species extrapolation methods, the AC-PBPK model can predict the in vivo exposure in human body and other species. It has reported that AC compatibility reduces the apparent permeability coefficient of transmembrane transport and other kinds of CYP enzymes and transporters participated in the in vivo process, which induced the relative error between the simulated and observed values. More factors will also be considered in the subsequent simulation.
Conclusion
In this study, amounts of measured physiology and PK parameters about AC were collected in the first stage, and the suitable fit values were added for lack parameters. After intravenous and oral PBPK model were established and optimized, the accuracy of AC-PBPK model was verified with multiple dose and administration mode. Further, the simulation by established AC-PBPK model exhibited a more effective and safe administration regimen for postprandial oral administration rather than 24-hour interval oral administration. After that, PBPK models of GA and PAE were constructed and introduced to the model of perpetrator drugs. The DDI models could accurately predict the in vivo process and tissue distribution of AC and the DDI effect of GA and PAE, which reveal one possible mechanism of detoxification of AC after combined therapy. To summarize, the development of AC-PBPK model provide an appropriate tool for in vivo AC studies, new safety evaluation method for compatibility of medicines and extra support for AC mechanism research.
Author Statements
Highlights
Aconitine has an impact on the process of several cardiac and nervous disease.
In vivo process of aconitine was simulated by established AC-PBPK model accurately.
Established PBPK model described drug-drug interactions correctly.
We discuss the importance and new applications of AC-PBPK model.
Acknowledgements
This project was supported by Major Program of National Natural Science Foundation of China (Nos. 82192910, 82192911). The National Natural Science Foundation of China (NO. 81873063) and Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (NO. ZYYCXTD-C-202009).
Authors' Contributions
YX. G designed this study; YX. G and Z. Z performed most of the experiments, analyzed the data and drafted the manuscript; XM. Z, JY. Y, G. J, ZC. M and YG. W assisted with in the experiments; Y. G reviewed the manuscript.
Conflicts of Interest
There are no conflicts of interest to declare.
References
- Wang H, Liu Y, Guo Z, Wu K, Zhang Y, et al. Aconitine induces cell apoptosis via mitochondria and death receptor signaling pathways in hippocampus cell line. Res Vet Sci. 2022; 143: 124-33.
- Xia Q, Gao S, Rapael Gnanamuthu SR, Zhuang K, Song Z, et al. Involvement of Nrf2-HO-1/JNK-Erk Signaling Pathways in Aconitine-Induced Developmental Toxicity, Oxidative Stress, and ROS-Mitochondrial Apoptosis in Zebrafish Embryos. Frontiers in pharmacology. 2021; 12: 642480.
- Karturi SP, Gudmundsson H, Akhtar M, Jahangir A, Choudhuri I. Spectrum of cardiac manifestations from aconitine poisoning. HeartRhythm Case Rep. 2016; 2: 415-20.
- Sun S, Chen Q, Ge J, Liu X, Wang X, et al. Pharmacokinetic interaction of aconitine, liquiritin and 6-gingerol in a traditional Chinese herbal formula, Sini Decoction. Xenobiotica. 2018; 48: 45-52.
- Yang L, Wang Y, Xu H, Huang G, Zhang Z, et al. Panax ginseng Inhibits Metabolism of Diester Alkaloids by Downregulating CYP3A4 Enzyme Activity via the Pregnane X Receptor. Evid Based Complement Alternat Med. 2019; 2019: 3508658.
- Zhang M, Peng CS, Li XB. Human intestine and liver microsomal metabolic differences between C19-diester and monoester diterpenoid alkaloids from the roots of Aconitum carmichaelii Debx. Toxicol In Vitro. 2017; 45: 318-33.
- Chen R, Ning Z, Zheng C, Yang Y, Zhang C, et al. Simultaneous determination of 16 alkaloids in blood by ultrahigh-performance liquid chromatography-tandem mass spectrometry coupled with supported liquid extraction. J Chromatogr B Analyt Technol Biomed Life Sci. 2019; 1128: 121789.
- Haron MH, Avula B, Ali Z, Chittiboyina AG, Khan IA, et al. Assessment of Herb-Drug Interaction Potential of Five Common Species of Licorice and Their Phytochemical Constituents. J Diet Suppl. 2022; 1-20.
- Chen Q, Yin C, Li Y, Yang Z, Tian Z. Pharmacokinetic interaction between peimine and paeoniflorin in rats and its potential mechanism. Pharm Biol. 2021; 59: 129-33.
- Sun H, Wang J, Lv J. Effects of glycyrrhizin on the pharmacokinetics of paeoniflorin in rats and its potential mechanism. Pharm Biol. 2019; 57: 550-4.
- Li J, Zhang SH, He D, Wang JF, Li JQ. Paeoniflorin reduced the cardiotoxicity of aconitine in h9c2 cells. J Biol Regul Homeost Agents. 2019; 33: 1425-36.
- Li Z, Yan M, Cao L, Fang P, Guo Z, et al. Glycyrrhetinic Acid Accelerates the Clearance of Triptolide through P-gp In Vitro. Phytother Res. 2017; 31: 1090-6.
- Madny MA, Deshpande P, Tumuluri V, Borde P, Sangana R. Physiologically Based Biopharmaceutics Model of Vildagliptin Modified Release (MR) Tablets to Predict In Vivo Performance and Establish Clinically Relevant Dissolution Specifications. AAPS PharmSciTech. 2022; 23: 108.
- Wang Z, Chan ECY. Physiologically Based Pharmacokinetic Modelling to Investigate Baricitinib and Tofacitinib Dosing Recommendations for COVID-19 in Geriatrics. Clin Pharmacol Ther. 2022.
- Wisniowska B, Giebultowicz J, Piotrowski R, Kulakowski P, Polak S. Development and Performance Verification of the PBPK Model for Antazoline and Its Metabolite and Its Utilization for Pharmacological Hypotheses Formulating. Pharmaceuticals (Basel). 2022; 15.
- Albrecht M, Kogan Y, Kulms D, Sauter T. Mechanistically Coupled PK (MCPK) Model to Describe Enzyme Induction and Occupancy Dependent DDI of Dabrafenib Metabolism. Pharmaceutics. 2022; 14.
- Tang L, Gong Y, Lv C, Ye L, Liu L, et al. Pharmacokinetics of aconitine as the targeted marker of Fuzi (Aconitum carmichaeli) following single and multiple oral administrations of Fuzi extracts in rat by UPLC/MS/MS. Journal of ethnopharmacology. 2012; 141: 736-41.
- Sun HY, Li Q, Chen W, Geng LL, Li X, et al. Pharmacokinetic analysis of alpha and beta epimers of glycyrrhetinic acid in rat plasma: Differences in singly and combined administrations. Yao Xue Xue Bao. 2012; 47: 94-100.
- Fan YF, Xie Y, Liu L, Ho HM, Wong YF, et al. Paeoniflorin reduced acute toxicity of aconitine in rats is associated with the pharmacokinetic alteration of aconitine. Journal of ethnopharmacology. 2012; 141: 701-8.
- He Y, Wei Z, Xie Y, Yi X, Zeng Y, et al. Potential synergic mechanism of Wutou-Gancao herb-pair by inhibiting efflux transporter P-glycoprotein. J Pharm Anal. 2020; 10: 178-86.
- Tazawa T, Zhao HQ, Li Y, Meselhy MR, Nakamura N, et al. A new enzyme immunoassay for aconitine and its application to quantitative determination of aconitine levels in plasma. Biological & pharmaceutical bulletin. 2003; 26: 1289-94.
- Ye L, Yang X, Yang Z, Gao S, Yin T, et al. The role of efflux transporters on the transport of highly toxic aconitine, mesaconitine, hypaconitine, and their hydrolysates, as determined in cultured Caco-2 and transfected MDCKII cells. Toxicol Lett. 2013; 216: 86-99.
- Yang C, Li Z, Zhang T, Liu F, Ruan J, et al. Transcellular transport of aconitine across human intestinal Caco-2 cells. Food Chem Toxicol. 2013; 57: 195-200.
- Wu J, Lin N, Li F, Zhang G, He S, et al. Induction of P-glycoprotein expression and activity by Aconitum alkaloids: Implication for clinical drug-drug interactions. Scientific reports. 2016; 6: 25343.
- Mattarello MJ, Benedini S, Fiore C, Camozzi V, Sartorato P, et al. Effect of licorice on PTH levels in healthy women. Steroids. 2006; 71: 403-8.
- Zhang L, Wei W. Anti-inflammatory and immunoregulatory effects of paeoniflorin and total glucosides of paeony. Pharmacol Ther. 2020; 207: 107452.
- Zhu L, Wu J, Zhao M, Song W, Qi X, et al. Mdr1a plays a crucial role in regulating the analgesic effect and toxicity of aconitine by altering its pharmacokinetic characteristics. Toxicology and applied pharmacology. 2017; 320: 32-9.