
Research Article
J Bacteriol Mycol. 2023; 10(3): 1213.
Screening Targets for Diagnosis and Treatment of Cognitive Dysfunction in Stroke through Transcriptome Combined with Machine Learning
Zhibin Chen1*#; Junxiong Li2#; Yangbo Hou1#; Qiaoyan Zhu1*; Zhizhen Shi1*
¹Department of Neurology, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
²Department of Acupuncture, Huadong Hospital Affiliated to Fudan University, Shanghai 20040, China
*Corresponding author: Zhibin Chen Department of Neurology, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China. Email: 13207919029@163.com
#These authors have been contributed equally to this author.
Received: October 25, 2023 Accepted: November 27, 2023 Published: December 04, 2023
Abstract
Post-Stroke Cognitive Impairment (PSCI) is one of the major complications after stroke. The evaluation of PSCI usually depends on neuropsychology tests, but the results of these tests are subjective and inaccurate. Need to find more objective indicators as identification markers of PSCI. In this study, we use machine learning to find biomakers of PSCI, and established regulatory networks at transcriptional level. Several gene such as ORC1, TOMM40L and SHISAL2A are identified biomakers, and several miRNA such as hsa-mir-130b-3p and hsa-mir-484 are interacted most tight with this biomakers genes. The results of this study help to better distinguish patients with PS and PSCI in clinical practice, and identifying relevant biomarker genes and miRNAs that can serve as potential therapeutic sites.
Keywords: PSCI; Machine learning
Introduction
Post-Stroke Cognitive Impairment (PSCI) is a clinical syndrome characterized by varying degrees of cognitive impairment that occur within 3 months after a stroke [1]. It encompasses different types of cognitive impairments resulting from stroke events, such as multiple infarctions, infarctions in critical areas, subcortical infarctions, and cerebral hemorrhages [2]. PSCI can also include clinical subtypes where cognitive impairment worsens in other neurodegenerative diseases following a stroke event.Previous study has reported that patients with post-stroke cognitive impairment exhibit an 8% mortality rate within 1.5 years [3]. However, the mortality rate significantly rises to 50% when the condition progresses to late-stage post-stroke dementia. Due to advancements in sequencing technology, gene sequencing has become extensively utilized in disease research. Analyzing gene expression profiling in patients' peripheral blood holds great significance for early disease detection [4]. The development of disease classifiers based on patient gene expression data using machine learning methods has gained substantial attention recently. Machine learning techniques have already found widespread application in the clinical diagnosis of cardiovascular diseases, such as coronary artery calcification scoring. The integration of key mRNAs and traditional diagnostic methods shows promise in enhancing the latter's accuracy [5]. In this study, we obtained gene expression data sets from stroke patients and post-stroke cognitive impairment patients in the Gene Expression Omnibus (GEO) database. We utilized the XG-Boost machine learning algorithm to identify distinguishing feature genes. Subsequently, the gene expression profiles were tested in the collected clinical samples. The identified feature genes in this study have potential applications in diagnosis and as biomarkers.
Materials and Methods
Data Sources
We used bioinformatics and experimental methods to explore the biological characteristics of sepsis. First, we used the GEO query package of the R software (version 4.1.0, http://rproject.org/) to download the sample source from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/ ) database. The reliable sepsis expression profile GSE186798 are all from Homo sapiens. GSE186798 is based on GPL23038 and GPL23159. This data set contains 60 brain tissue, including 30 sepsis and 30 healthy controls.
Gene Ontology and Functional Enrichment Analysis
We conducted Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis (www.genome.jp/kegg/) to identify the biological functions of the genes. Significant pathways with a P-value less than 0.05 were ultimately selected.
Immune Infiltration Analysis
We employed the CIBERSORT algorithm to examine the connection between genes associated with diagnosis and the expression of immune cell-related markers. In particular, we calculated the relative proportions of various immune cell types in the significant samples (P<0.05) from the GSE186798 dataset. This analysis provided us with the abundance of 22 immune cell types and allowed us to determine the correlation between the diagnosis-related genes and the content of each immune cell type using the Spearman correlation coefficient. Additionally, we conducted Pearson correlation analysis using the GSE186798 dataset to evaluate the correlation between immune test sites and the diagnosis-related genes.
Identification of Transcription Factors and miRNAs
In order to better comprehend the major variations at the transcriptional level and gain insights into the crucial regulatory molecules, we investigated the interaction networks between Differentially Expressed Genes (DEGs) and microRNAs (miRNAs), as well as the interaction networks between Transcription Factors (TFs) and DEGs. In our analysis, we employed the NetworkAnalyst platform to identify TFs from the JASPAR database that displayed significant topological relevance and had a tendency to bind to the common DEGs. To construct the DEG-miRNA network, we utilized the TarBase and miRTarBase databases to extract miRNAs that were associated with the common DEGs, with a particular focus on topological analysis.
Evaluation of Applicant Drugs
In this analysis, the Protein–Drug Interaction (PDI) and identified pharmacological molecules were predicted by using the common DEGs. The web portal od Enrichr and the Drug Signatures Database (DSigDB) were used to analyze the drug moleculars based on the DEGs . Enrichr (http://amp.pharm. mssm.edu/Enrichr) contains a large collection of diverse gene set libraries available for analysis and download, which can be used to explore gene-set enrichment across a genome-wide scale (39). DSigDB is a new gene set resource for gene set enrichment analysis, which related drugs/compounds and their target genes. The DSigDB database was accessed through Enrichr under the Diseases/Drugs function.
Results
PS and PSCI Has no Significant Different on Gene Expression Model
Principal Component Analysis (PCA), is a dimensionality reduction method that is often used to reduce the dimensionality of large data sets, by transforming a large set of variables into a smaller one that still contains most of the information in the large set [6,7]. However, the disadvantage of PCA is that the data has not passed the Permutation test, Permutational Multivariate Analysis of Variance (PERMANOVA) uses the Distance matrix (such as Euclidean distance and Bray Curtis distance) to decompose the total variance, analyze the explanatory power of different grouping factors or different environmental factors on sample differences, and use Permutation test to analyze the statistical significance of each variable interpretation [1,2]. In this study, PCA and PERMANOVA were used to determine whether there was a difference in gene expression between PS and PSCI. From the figure, it can be seen that the sample distribution of PS and PSIC is uniform and there is no giant difference, and the P-value obtained by the PERMANOVA algorithm is 0.978, which indicates that the gene expression pattern bwtween PS and PSIC has no significant differences (Figure 1A).
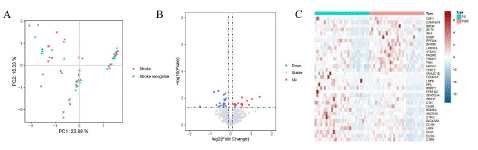
Figure 1: A) PCA and PERMANOVA of PS and PSIC. B) Volcano plot indicates significant diferntially expressed genes, with red dots indicating high expression and blue dots indicating low expression. C) The heat map of 34 diferntially expressed genes.
Identifcation of Diferentially Expressed Genes between PS and PSIC
We obtained 30 PS and 30 PSIC patients from GSE186798. A total of 34 diferntially expressed genes were identifed based on the cutof criteria of |log2 (fold change) |>0.1 and false discovery rate (P value)<0.05 using R package “Limma”(Figure 1B) . And the expression level of 34 diferntially expressed genes in 60 samples are displayed in the form of a heat map using R package “pheatmap” (Figure 1C).
KEGG and GO Enrichment Analysis for DEGs
To further clarify the main biological functions of the 34 DEGs, we performed KEGG and GO functional analysis of the 34 gene by DAVID (https://david.ncifcrf.gov/)(Figure 2). The KEGG enrichment analysis showed that the 34 different gene are enriched at pathways such as Amino sugar and nucleotide sugar metabolism, Hematopoietic cell lineage, Osteoclast differentiation, andGO analysis enrichedat chitinase activity, Chitin catabolic process, chitin binding, positive regulation of protein tyrosine kinase activity and so on.

Figure 2: KEGG and GO enrichment analysis of the 34 gene (Counts represent genes enriched in pathway).
The XGBoost model completely groups PS and PSCI
Machine learning is generally divided into supervised learning and unsupervised learning [8]. Supervised learning applies classification tasks and regression tasks, where the predicted labels for classification tasks are discrete, while the predicted labels for regression tasks are continuous [9,10]. XGBoost is an integrated Tree model, which uses the sum of K CART regression trees to predict the sample values as the prediction result.XGBoost (eXtreme Gradient Boosting) is a popular machine learning algorithm, and it is wide used in classification [11,12]. We use 60 samples of PS and PSCI for XGBoost model training, and the score of most important top ten gene identified by XGBoost is showed in Figure 3A.
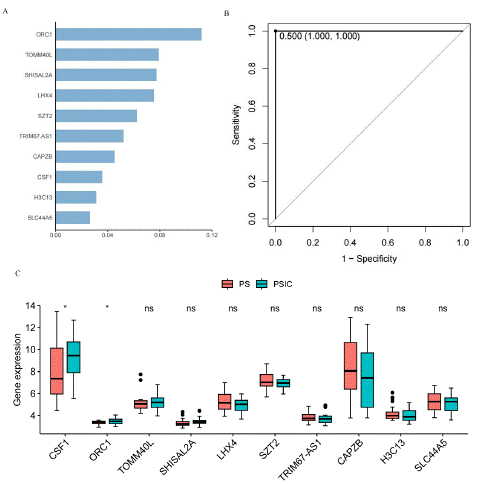
Figure 3: XGBoost analysis of PS and PSIC. A) The most important top 10 gene of XGBoost model. B) ROC curve of XGBoost model. C) Abundances of mRNA levels between PS and PSIC samples (Wilcxo test, *p<0.05, ns, not significance).
They are ORC1, TOMM40L, SHISAL2A, LHX4, SZT2, TRIM67. AS1, CAPZB, CSF1, H3C13 and SLC44A5. In addition, ROC curve was used to test the correct classification ability of the xgboost model, the AUC of this model is 1, which means this model has completely predicted success [13,14]. The mRNA levels of XGBoost related genes (log2-transformed) were compared between PS and PSIC samples with wilcox test. Among these ten genes, the expression levels of CSF1 and ORC1 significantly increased in PSIC. Therefore, these ten genes are of great significance for disease classification, and CSF1 and ORC1 can serve as biomarkers.
Construction of Regulatory Networks at Transcriptional Level
To identify substantial changes happening at the transcriptional level and get insights into the common DEGs, a network-based approach was employed to decode the regulatory TFs and miRNAs [15,16]. The DEG–TFs interactions network was identified by using TarBase and miRTarBase bases and displayed in [17] Figure 4. Circles represented common DEGs, while diamonds were TFs. The size of the circular or rhombus node depends on the degree of the node. The degree of a node is the number of connections the node has with other nodes in the network. Nodes with a higher degree are considered as important hubs of the network. From the Figure 4, TOMM40L, CSF1, SZT2, ORC1, HIST2H3D and LHX4 were more among more highly expressed DEGs as these genes have a higher degree in the TF-gene interactions network. TFs such as ZNF394, ELK1, SP1, ATF1, ZNF175, HBP1, IRF1, TRIM24 and MLLT1 were more significant than others as presented in the same figure 4. Again, the Figure 5 represented the interactions of miRNAs regulators with common DEGs. In the Figure 5, red squares represented miRNAs, while blue circles represented DEGs. Our results showed that CAPZB, ORC1, TOMM40L, SZT2, CSF1, LHX4 and HIST2H3D were the hub genes of this network, with the genes most involved in miRNAs. Besides, the significant hub miRNAs were detected from the miRNAs-gene interaction network, namely hsa-mir-130b-3p, hsa-mir-484, hsa-mir-128-3p, hsa-mir-92b-3p, hsa-mir-661, hsa-mir-939-3p, hsa-mir-6849-3p, hsa-mir-34a-5p, hsa-mir-6894-5p and hsa-mir-130a-3p.
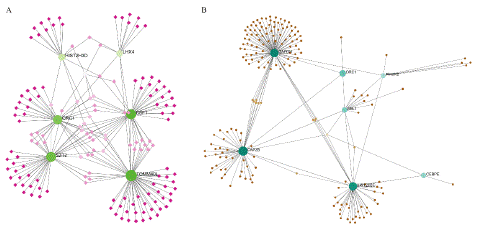
Figure 4: Interactions network A) DEG–TFs interactions network. B) DEG–miRNAs interactions network.
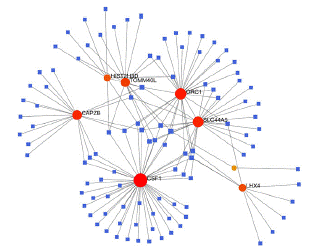
Figure 5: Interactions of miRNAs regulators with common DEGs(Red squares represented miRNAs, blue circles represented DEGs).
Identification of Candidate Drugs and Target–Chemical Interaction in PSCI
A chemical–protein interaction network is an important research tool for understanding the function of proteins, which is helpful for advancing drug discovery [18,19]. In the aspects of common DEGs as potential drug targets in PSCI, the candidate drugs were identified by using Enrichr based on transcriptome signatures from the DSigDB database [20,21]. The top 10 drug molecules selected based on p-value were considered as potential compounds that could be used for PSCI treatment and subsequent analysis. These 10 possible drug molecules included (+)-JQ1 compound, Aflatoxin B1, Methyl Methanesulfonate, Calcitriol, Cyclosporine, Tetrachlorodibenzodioxin, Silicon Dioxide, Testosterone, resveratrol,Copper Sulfate as shown in Figure 6.
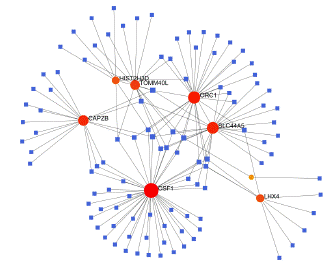
Figure 6:
Discussion
Early recognition and prompt treatment of PSCI are crucial for improving patient outcomes.This study aims to investigate the molecular dysregulation mechanisms associated with PSCI by leveraging bioinformatics analysis of PSCI-related sequencing data. By subjecting the intersected genes to enrichment analyses, we aimed to determine whether specific biological pathways were overrepresented among these genes. The GO and KEGG enrichment analysis showed that multiple immune-related pathways that were significantly enriched among the intersected genes. The KEGG enrichment analysis showed that the P values of signaling pathways such as Amino sugar and nucleotide sugar metabolism, Hematopoietic cell lineage, Osteoclast differentiation. The GO analysis showed that P values of signaling pathways such as chitinase activity, Chitin catabolic process, chitin binding, positive regulation of protein tyrosine kinase activity and so on.
XGBoost is a popular machine learning algorithm that is widely used for regression, classification, and ranking tasks [22]. It is an implementation of gradient boosting trees, which are a type of ensemble learning algorithm [23]. XGBoost is known for its high performance and efficiency, and it has won numerous machine learning competitions on Kaggle [24]. It combines the advantages of boosting algorithms with efficient implementation techniques to achieve high accuracy and speed [25]. In this study, XGBoost was used to identify the genes, such as ORC1, TOMM40L, SHISAL2A, LHX4, SZT2, TRIM67.AS1, CAPZB, CSF1, H3C13, and SLC44A5.
To explore the transcriptional regulation of sepsis by commonly observed Differentially Expressed Genes (DEGs), we utilized web tools to investigate the interactions among Transcription Factors (TFs), microRNAs (miRNAs), and genes. Our result shows eight miRNAs, namely hsa-mir-130b-3p, hsa-mir-484, hsa-mir-128-3p, hsa-mir-92b-3p, hsa-mir-661, hsa-mir-939-3p, hsa-mir-6849-3p, hsa-mir-335-5p, were identified to be associated with the DEGs. Although many previous studies have suggested that these TFs and miRNAs may have important therapeutic effects, these analytical results require further experiments to confirm their validity and authenticity.
On the other hand, our analysis revealed the identification of key genes that appear to have a potential influence on the development of PSCI. The TOMM40L gene has been increasingly studied in relation to post-stroke cognitive impairment (PSCI). Several studies have suggested that variations in the TOMM40L gene may play a role in the development of PSCI [26]. In particular, a specific genotype of TOMM40L, known as the long poly-T variant, has been associated with an increased risk of PSCI. LHX4 (Lim-homeobox protein 4) is a gene that encodes a transcription factor implicated in brain development and function [27]. However, there was no report about the relationship between the LHX4 and the PSCI. SZT2 (Seizure Threshold 2) is a gene that has gained attention for its potential role in PSCI. An previous study conducted genetic analyses on a population of stroke patients and found that certain variants of the SZT2 gene were associated with an increased risk of PSCI [28]. Another study found that SZT2 gene expression was altered in the brains of individuals with PSCI compared to those without cognitive impairments after stroke [29]. The mechanisms by which SZT2 may contribute to PSCI are not yet fully understood. TRIM67.AS1 is a long non-coding RNA that has been implicated in a variety of biological processes and diseases, including Post-Stroke Cognitive Impairment (PSCI) [30]. Recent studies have identified dysregulation of TRIM67.AS1 expression in the brains of individuals with PSCI compared to those without cognitive impairments after a stroke. However, the specific role of TRIM67. AS1 in PSCI is not yet fully understood. CAPZB (Capping protein, muscle Z-line beta) is a gene that encodes a protein involved in the regulation of actin filaments, which play a crucial role in various cellular processes, including neuronal development and synaptic function. While research on the role of CAPZB in Post-Stroke Cognitive Impairment (PSCI) is limited, studies suggest its potential involvement in cognitive impairments after a stroke. In one study, the expression of CAPZB was found to be significantly altered in the brains of individuals with PSCI compared to those without cognitive impairments after a stroke [31]. The researchers proposed that dysregulation of CAPZB may disrupt actin dynamics, which could contribute to the synaptic dysfunction and neuronal damage observed in PSCI [32]. CSF1 (Colony Stimulating Factor 1) is a gene that encodes a cytokine known as CSF1 or Macrophage Colony-Stimulating Factor (M-CSF). CSF1 plays a critical role in the regulation and differentiation of macrophages and microglia, which are key immune cells in the central nervous system [33]. While the direct role of CSF1 in Post-Stroke Cognitive Impairment (PSCI) is not yet fully understood, emerging evidence suggests its involvement in the neuroinflammatory processes that contribute to cognitive impairments after a stroke. SLC44A5 plays a role in neuronal development, neurite outgrowth, and synaptic function. Alterations in SLC44A5 expression or function may impact neuronal connectivity and synaptic plasticity, which are essential for normal cognitive function, while there is no direct evidence linking SLC44A5 to PSCI, studies in other neurological disorders have shed some light on its potential involvement in cognitive impairments. For example, altered SLC44A5 expression has been observed in the brains of individuals with Alzheimer's disease and schizophrenia, both of which are associated with cognitive deficits. In this study, we focused on exploring the potential diagnostic value of PSCI-related genes. Through this research, we anticipate that we will gain a more comprehensive of the mechanism of PSCI, potentially leading to improved prognosis, tailored treatment strategies, and better patient outcomes. It will contribute to advancing the field of precision medicine in PSCI management.
Author Statements
Disclosure of Conflicting Interests
We hereby state that there are no potential conflicts of interest.
Funding Sources
This research was supported by the Funding source: Medical Alliance Research Project of Shanghai Sixth People's Hospital (21-LY-03).
Acknowledgments
The authors would like to thank Prof. Xirong Guo and Xingyun Wang Ph.D (Hongqiao International Institute of Medicine, Tongren Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200240, China), for critically reviewing the manuscript.
References
- Chi NF, Chao SP, Huang LK, Chan L, Chen YR, Chiou HY, et al. Plasma amyloid beta and tau levels are predictors of post-stroke cognitive impairment: A longitudinal study. Front Neurol. 2019; 10: 715.
- Ma C, Wang D, Li X, Feng Q, Liu Y, Hong Z, Chen L. Multivariate logistic regression analysis of clinical characteristics and risk factors of cognitive impairment after cerebral ischemic stroke: implications for clinical treatment. Ann Transl Med. 2023; 11: 318.
- Treadaway C, Seckam A, Fennell J, Taylor A. HUG: A compassionate approach to designing for wellbeing in dementia care. Int J Environ Res Public Health. 2023; 20: 4410.
- Shou J, Bull CM, Li L, Qian HR, Wei T, Luo S, et al. Identification of blood biomarkers of rheumatoid arthritis by transcript profiling of peripheral blood mononuclear cells from the rat collagen-induced arthritis model. Arthritis Res Ther. 2006; 8: R28.
- Shang Z, Sun J, Hui J, Yu Y, Bian X, Yang B, et al. Construction of a support vector machine-based classifier for pulmonary arterial hypertension patients. Front Genet. 2021; 12: 781011.
- Xie J, Zhu G, Gao M, Xi J, Chen G, Ma X, et al. An artemisinin derivative ART1 induces ferroptosis by targeting the HSD17B4 protein essential for lipid metabolism and directly inducing lipid peroxidation. CCS Chem. 2022; 4: 304-17.
- Sun Z, Wang X, Wang J, Wang J, Liu X, Huang R, et al. Key radioresistance regulation models and marker genes identified by integrated transcriptome analysis in nasopharyngeal carcinoma. Cancer Med. 2021; 10: 7404-17.
- Patel V, Khan MN, Shrivastava A, Sadiq K, Ali SA, Moore SR, et al. Artificial intelligence applied to gastrointestinal diagnostics: a review. J Pediatr Gastroenterol Nutr. 2020; 70: 4-11.
- Cheng L, Qiu Y, Schmidt BJ, Wei GW. Review of applications and challenges of quantitative systems pharmacology modeling and machine learning for heart failure. J Pharmacokinet Pharmacodyn. 2022; 49: 39-50.
- Pawar S, San O, Vedula P, Rasheed A, Kvamsdal T. Multi-fidelity information fusion with concatenated neural networks. Sci Rep. 2022; 12: 5900.
- Baskor A, Tok YP, Mesut B, Özsoy Y, Uçar T. Estimating the optimal dexketoprofen pharmaceutical formulation with machine learning methods and statistical approaches. Healthc Inform Res. 2021; 27: 279-86.
- Ma J, Bo Z, Zhao Z, Yang J, Yang Y, Li H, et al. Machine learning to predict the response to lenvatinib combined with transarterial chemoembolization for unresectable hepatocellular carcinoma. Cancers (Basel). 2023; 15.
- Zhang J, Jun T, Frank J, Nirenberg S, Kovatch P, Huang KL. Prediction of individual COVID-19 diagnosis using baseline demographics and lab data. Sci Rep. 2021; 11: 13913.
- Beuque M, Magee DR, Chatterjee A, Woodruff HC, Langley RE, Allum W, et al. Automated detection and delineation of lymph nodes in haematoxylin & eosin stained digitised slides. J Pathol Inform. 2023; 14: 100192.
- Lu L, Liu LP, Gui R, Dong H, Su YR, Zhou XH, et al. Discovering common pathogenetic processes between COVID-19 and sepsis by bioinformatics and system biology approach. Front Immunol. 2022; 13: 975848.
- Zhang W, Liu L, Xiao X, Zhou H, Peng Z, Wang W, et al. Identification of common molecular signatures of SARS-CoV-2 infection and its influence on acute kidney injury and chronic kidney disease. Front Immunol. 2023; 14: 961642.
- Xia J, Chen S, Li Y, Li H, Gan M, Wu J, et al. Immune response is key to genetic mechanisms of SARS-CoV-2 infection with psychiatric disorders based on differential gene expression pattern analysis. Front Immunol. 2022; 13: 798538.
- Yoo HY, Lee KC, Woo JE, Park SH, Lee S, Joo J, et al. A genome-wide association study and machine-learning algorithm analysis on the prediction of facial phenotypes by genotypes in Korean women. Clin Cosmet Investig Dermatol. 2022; 15: 433-45.
- Nashiry MA, Sumi SS, Sharif Shohan MU, Alyami SA, Azad AKM, Moni MA. Bioinformatics and system biology approaches to identify the diseasome and comorbidities complexities of SARS-CoV-2 infection with the digestive tract disorders. Brief Bioinform. 2021; 22.
- Elango R, Banaganapalli B, Mujalli A, AlRayes N, Almaghrabi S, Almansouri M, et al. Potential biomarkers for Parkinson disease from functional enrichment and bioinformatic analysis of global gene expression patterns of blood and substantia nigra tissues. Bioinform Biol Insights. 2023; 17: 11779322231166214.
- Li P, Li T, Zhang Z, Dai X, Zeng B, Li Z, et al. Bioinformatics and system biology approach to identify the influences among COVID-19, ARDS and sepsis. Front Immunol. 2023; 14: 1152186.
- Sunarya U, Sun Hariyani Y, Cho T, Roh J, Hyeong J, Sohn I, et al. Feature analysis of smart shoe sensors for classification of gait patterns. Sensors (Basel). 2020; 20.
- Abe D, Inaji M, Hase T, Takahashi S, Sakai R, Ayabe F, et al. A prehospital triage system to detect traumatic intracranial hemorrhage using machine learning algorithms. JAMA Netw Open. 2022; 5: e2216393.
- Hsu A, Wang X, Tan J, Toh W, Goyal N. Predicting European cities’ climate mitigation performance using machine learning. Nat Commun. 2022; 13: 7487.
- Liu Z, Zhou T, Han X, Lang T, Liu S, Zhang P, et al. Mathematical models of amino acid panel for assisting diagnosis of children acute leukemia. J Transl Med. 2019; 17: 38.
- Cardoso R, Lemos C, Oliveiros B, Almeida MR, Baldeiras I, Pereira CF, et al. APOEvarepsilon4-TOMM40L haplotype increases the risk of mild cognitive impairment conversion to Alzheimer’s disease. J Alzheimers Dis. 2020; 78: 587-601.
- Herrera-Campos AB, Zamudio-Martinez E, Delgado-Bellido D, Fernández-Cortés M, Montuenga LM, Oliver FJ et al. Implications of hyperoxia over the tumor microenvironment: an overview highlighting the importance of the immune system. Cancers (Basel). 2022; 14.
- Pizzino A, Whitehead M, Sabet Rasekh P, Murphy J, Helman G, Bloom M, et al. Mutations in SZT2 result in early-onset epileptic encephalopathy and leukoencephalopathy. Am J Med Genet A. 2018; 176: 1443-8.
- Hong SY, Yang JJ, Li SY, Lee IC. A wide spectrum of genetic disorders causing severe childhood epilepsy in Taiwan: A case series of ultrarare genetic cause and novel mutation analysis in a pilot study. J Pers Med. 2020; 10.
- Mehler MF. Introduction: epigenetics and neuropsychiatric diseases. Neurobiol Dis. 2010; 39: 1-2.
- Brambilla S, Guiotto M, Torretta E, Armenia I, Moretti M, Gelfi C, et al. Human platelet lysate stimulates neurotrophic properties of human adipose-derived stem cells better than Schwann cell-like cells. Stem Cell Res Ther. 2023; 14: 179.
- Pinosanu LR, Capitanescu B, Glavan D, Godeanu S, Cadenas IFN, Doeppner TR, et al. Neuroglia cells transcriptomic in brain development, aging and neurodegenerative diseases. Aging Dis. 2023; 14: 63-83.
- Foulds N, Pengelly RJ, Hammans SR, Nicoll JA, Ellison DW, Ditchfield A, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia caused by a novel R782G mutation in CSF1R. Sci Rep. 2015; 5: 10042.